New Packages on CRAN: tidycwl and biocompute
On the journey of achieving reproducibility in genomic data analysis projects, one often faces challenges with documenting workflows and computations systematically. To provide one way for tackling these problems, we (me and my colleagues) have recently released two new R packages — tidycwl and biocompute — to CRAN.
tidycwl
As the name implies, the package tidycwl aims at offering a native toolchain for R to analyze tools and workflows written in the Common Workflow Language (CWL), while following the tidyverse design principles. What it does is not complicated at all: reading the raw CWL workflows encoded in JSON or YAML, parsing the workflow elements into data frames, lists, or directed graph representations, and visualizing the workflows.
For example, to visualize a CWL BWA + GATK 4 whole genome sequencing workflow, a few pipes get the job done:
library("tidycwl")
flow <- system.file("cwl/sbg/workflow/gatk4-wgs.json", package = "tidycwl") %>%
read_cwl_json()
get_graph(
flow %>% parse_inputs(),
flow %>% parse_outputs(),
flow %>% parse_steps()
) %>% visualize_graph(palette = c("#E69F00", "#56B4E9", "#009E73"))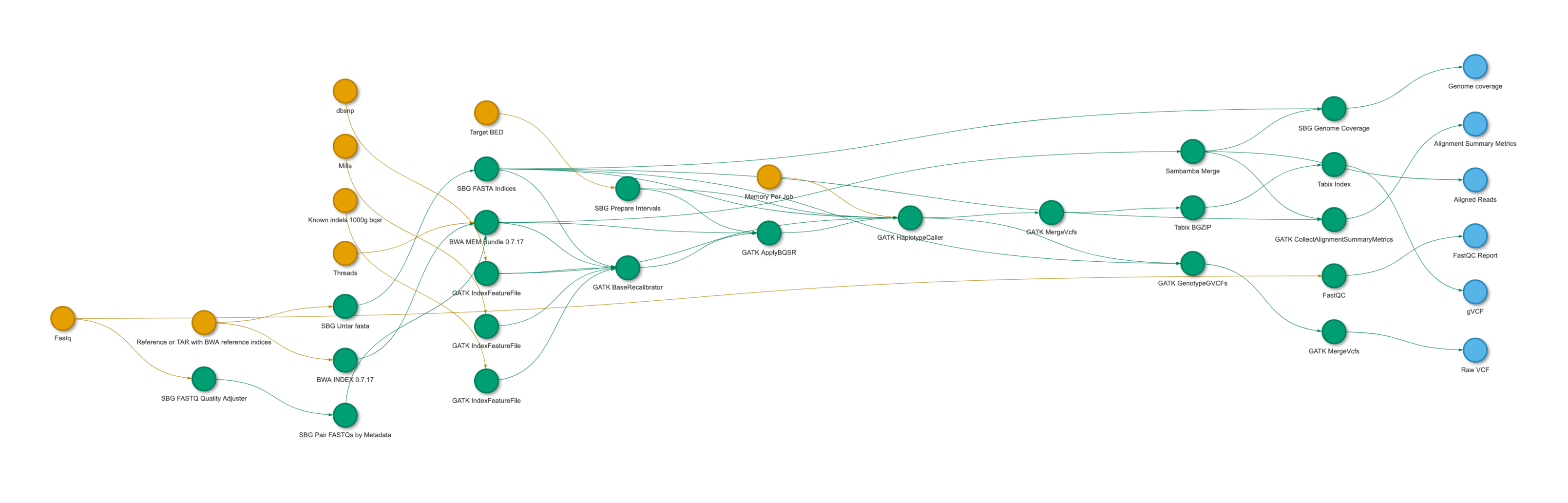
A tidycwl visualization of the GATK 4 WGS workflow. You can see the interactive version here.
The number of workflows written in the Common Workflow Language is growing every day. I believe the workflows themselves are becoming a valuable data type that can be displayed and analyzed by the data science community. My hope for tidycwl is to help people achieve these goals reliably without hassles at the toolchain level.
biocompute
The BioCompute Object (BCO) is an emerging open standard for documenting and sharing next-generation sequencing (NGS) data analyses. The standard has the potential to lower the burden for scientists to share, communicate, and review such computations across organizations, thus improve the speed of discovery, promote computational reproducibility, and in general, encourage open science. For more details about the BioCompute project, you can check out the excellent introductory blog post written by my colleagues.
The R package biocompute offers an implementation of the BioCompute Object specification. With the package, users can compose, validate, convert, and export BioCompute Objects in R with a functional API.
Thanks to the awesome infrastructure built by the R community in this decade, we can accomplish interoperability-related missions such as JSON schema validation or exporting to Word documents without reinventing many wheels. Let’s keep making it better.